






3-е изд., испр. - М.: Высшая школа, 2001 - 512 с., 319 с.
Учебник составлен в соответствии с программой по физической химии.
В первой книге подробно изложены следующие разделы курса: квантовомеханические основы теории химической связи, строение атомов и молекул, спектральные методы исследования молекулярной структуры, феноменологическая и статистическая термодинамика, термодинамика растворов и фазовых равновесий.
Во второй части раздела курса физической химии электрохимия, химическая кинетика и катализ излагаются на основе представлений, развитых в первой части книги, - строение вещества и статистическая термодинамика. В разделе `Катализ` отражены кинетика гетерогенных и диффузионных процессов, термодинамика адсорбции и вопросы реакционной способности.
Для студентов вузов, обучающихся по химико-технологическим специальностям.
Книга 1.
Формат: djvu
Размер: 11,2 Мб
Скачать: drive.google
Книга 2.
Формат: djvu
Размер: 7 Мб
Скачать: drive.google
ОГЛАВЛЕНИЕ Книга 1.
Предисловие. 3
Введение 6
Раздел первый. Квантовомеханическое обоснование теории строения молекул и
химической связи
Г л а в а 1. Строение атома 9
§ 1.1. Квантовомеханические особенности микрочастиц 9
§ 1.2. Водородоподобный атом 11
§ 1.3. Атомные орбитали водородоподобного атома 14
§ 1.4. Спин электрона 21
§ 1.5. Многоэлектронные атомы 23
§ 1.6. Принцип Паули 26
§ 1.7. Электронные конфигурации атомов 28
Г л а в а 2. Молекулы. Теоретические методы, применяемые при изучении
строения молекул и химической связи 34
§ 2.1. Молекула. Потенциальная поверхность. Равновесная конфигурация 34
§ 2.2. Теория химической связи и ее задачи. Уравнение Шредингера для молекул 39
§ 2.3. Вариационный метод решения уравнения Шредингера 42
§ 2.4. Два основных метода теории строения молекул. Метод валентных связей и
метод молекулярных орбиталей 44
§ 2.5. Основные идеи метода молекулярных орбиталей 49
§ 2.6. Приближенное описание молекулярной орбитали в методе МО ЛКАО 50
§ 2.7. Молекула Щ в методе МО ЛКАО. Расчет энергии и волновой функции по
вариационному методу 53
§ 2.8. Молекула Н в методе МО ЛКАО. Ковалентная связь 58
Г л а в а 3. Двухатомные молекулы в методе МО ЛКАО 62
§ 3.1. Молекулярные орбитали гомонуклеарных двухатомных молекул 62
§ 3.2. Электронные конфигурации и свойства гомонуклеарных молекул, образованных
атомами элементов первого и второго периодов 65
§ 3.3. Гетеронуклеарные двухатомные молекулы 73
§ 3.4. Полярная связь. Электрический дипольный момент молекулы 78
§ 3.5. Насыщаемость ковалентной связи 81
§ 3.6. Донорно-акцепторная связь 82
§ 3.7. Ионная связь. Степень полярности химической связи 84
Г л а в а 4. Многоатомные молекулы в методе МО 88
§ 4.1. Молекулярные орбитали в многоатомных молекулах. Симметрия орбиталей.
Делокализованные и локализованные орбитали. Молекула НгО 88
§ 4.2. Описание молекулы метана. Делокализованные и локализованные МО.
Гибридизация орбиталей 95
§ 4.3. О предсказании равновесных конфигураций молекул 99
§ 4.4. Нежесткие молекулы 101
§ 4.5. Молекулы с кратными связями в методе МО ЛКАО 104
§ 4.6. Метод Хюккеля 108
§ 4.7. Описание ароматических систем в методе МОХ 110
§ 4.8. Химическая связь в координационных соединениях. Теория поля лигандов 117
§ 4.9. Ионная связь в кристалле 126
Г л а в а 5. Межмолекулярное взаимодействие 129
§ 5.1. Силы Ван-дер-Ваальса. Другие виды неспецифического взаимодействия 129
§ 5.2. Водородная связь 136
Раздел второй. Спектральные методы исследования строения и энергетических
состояний молекул
Г л а в а 6. Общие сведения о молекулярных спектрах. Элементы теории
молекулярных спектров 141
§ 6.1. Внутримолекулярное движение и электромагнитный спектр. 141
§ 6.2. Молекулярные спектры испускания, поглощения и комбинационного рассеяния.
Спектры ЭПР и ЯМР 145
§ 6.3. Вращательный спектр двухатомной молекулы (приближение жесткого ротатора)
150
§ 6.4. Колебательно-вращательный спектр двухатомной молекулы. Приближение
гармонического осциллятора 156
§ 6.5. Молекула - ангармонический осциллятор. Структура колебательного спектра
162
§ 6.6. Электронные спектры. Определение энергии диссоциации двухатомных молекул
169
§ 6.7. Вращательные спектры и строгие многоатомных молекул.... 171
§ 6.8. Колебания, спектр и строение многоатомных молекул 175
§ 6.9. Использование колебательных спектров для определения строения молекул 180
§ 6.10. Влияние межмолекулярного взаимодействия среды и агрегатного состояния на
колебательный спектр 183
Раздел третий. Химическая термодинамика
Г л а в а 7. Общие понятия. Первый закон термодинамики и его приложение 186
§ 7.1. Предмет и задачи химической термодинамики 186
§ 7.2. Основные понятия и определения химической термодинамики 188
§ 7.3. Первый закон термодинамики. Некруговые процессы 199
§ 7.4. Теплоемкость 202
§ 7.5. Влияние температуры на теплоемкость. Температурные ряды.. 208
§ 7.6. Квантовая теория теплоемкости кристаллического вещества 211
§ 7.7. Квантовостатистическая теория теплоемкости газообразного вещества 215
§ 7.8. Тепловые эффекты. Закон Гесса 217
§ 7.9. Применение закона Гесса к расчету тепловых эффектов 220
§ 7.10. Зависимость теплового эффекта от температуры. Уравнение Кирхгофа 227
Г л а в а 8. Второй закон термодинамики и ею приложение 235
§ 8.1. Самопроизвольные и несамопроизвольные процессы. Второй закон
термодинамики 235
§ 8.2. Энтропия 236
§ 8.3. Изменение энтропии в нестатических процессах 239
§ 8.4. Изменение энтропии как критерий направленности и равновесия в
изолированной «истеме 240
§ 8.5. Характеристические функции. Термодинамические потенциалы 241
§ 8.6. Критерии возможности самопроизвольного процесса и равновесия в закрытых
системах 249
§ 8.7. Изменение энтропии в некоторых процессах 251
§ 8.8. Энергия Гиббса смеси идеальных газов. Химический потенциал 261
§ 8.9. Общие условия химического равновесия 265
§ 8.10. Закон действующих масс. Константа равновесия для газофазных реакций 266
§ 8.11. Уравнение изотермы реакции 271
§ 8.12. Использование закона действующих масс для расчета состава равновесной
смеси 273
§ 8.13. Влияние температуры на химическое равновесие. Уравнение изобары реакции
282
§ 8.14. Интегральная форма зависимости изменения энергии Гиббса и константы
равновесия от температуры 284
§ 8.15. Химическое равновесие в гетерогенных системах 286
Г л а в а 9. Третий закон термодинамики и расчет химического равновесия 289
§ 9.1. Тепловая теорема Нернста. Третий закон термодинамики 289
§ 9.2. Расчет изменения стандартной энергии Гиббса и константы равновесия по
методу Темкина - Шварцмана 294
§ 9.3. Расчет изменения стандартной энергии Гиббса и константы равновесия с
помощью функций приведенной энергии Гиббса 297
§ 9.4. Адиабатические реакции 299
Г л а в а 10. Химическое равновесие в реальных системах 303
§ 10.1. Фугитивность и коэффициент фугитивности газов 303
§ 10.2. Расчет химического равновесия в реальной газовой системе при высоких
давлениях 312
§ 10.3. Расчет химического равновесия в системах, в которых одновременно
протекает несколько реакций 314
Г л а в а 11. Введение в статистическую термодинамику 320
§ 11.1. Статистическая физика и статистическая термодинамика.
Макроскопическое и микроскопическое описание состояния системы 320
§ 11.2. Микроскопическое описание состояния методом классической механики 323
§ 11.3. Микроскопическое описание состояния методом квантовой механики.
Квантовые статистики 324
§ 11.4. Два вида средних величин (микрокано -нические и канонические средние)
325
§ 11.5. Связь энтропии и статистического веса. Статистический характер второго
закона термодинамики 326
§ 11.6. Система в термостате. Каноническое распределение Гиббса. 330
§ 11.7. Сумма по состояниям системы и ее связь с энергией. Гельмгольца 335
§ 11.8. Сумма по состояниям частицы 337
§ 11.9. Выражение термодинамических функций через сумму по состояниям системы
340
§ 11.10. Сумма по состояниям системы одномерных гармонических осцилляторов.
Термодинамические свойства одноатомного твердого тела по теории Эйнштейна 343
§ 11.11. Квантовая статистика Больцмана. Закон Максвелла распределения молекул
по скоростям 346
§ 11.12. Статистики Ферми - Дирака и Бозе - Эйнштейна 352
§ 11.13.Общие формулы для вычисления термодинамических функций по молекулярным
данным 353
§ 11.14.Вычисление термодинамических функций идеального газа в предположении
жесткого вращения и гармонических колебаний молекул 357
Раздел четвертый. Растворы
Г л а в а 12. Общая характеристика растворов 365
§ 12.1. Классификация растворов 365
§ 12.2. Концентрация растворов 367
5 12.3. Специфика растворов. Роль межмолекулярного и химического взаимодействий,
понятие о сольватации 368
§ 12.4. Основные направления в развитии теории растворов 372
§ 12.5. Термодинамические условия образования растворов 374
§ 12.6. Парциальные молярные величины 375
§ 12.7. Основные методы определения парциальных молярных величин 379
§ 12.8. Парциальные и относительные парциальные молярные энтальпии 381
§ 12.9. Теплоты растворения и разбавления 382
§ 12.10.Термодинамические свойства идеальных жидких растворов 386
§ 12.11.3акон Рауля 390
§ 12.12. Температура кипения идеального раствора 392
§ 12.13.Температура замерзания идеального раствора 395
§ 12.14.0смотическое давление идеального раствора 397
§ 12.15.Неидеальные растворы 400
§ 12.16. Предельно разбавленные, регулярные и атермальные растворы 402
§ 12.17. Активность. Коэффициент активности. Стандартное состояние 404
§ 12.18.0смотический коэффициент 407
§ 12.19.Методы определения активностей 409
§ 12.20.Связь коэффициента активности и активности с термодинамическими
свойствами раствора и избыточные термодинамические функции 412
Раздел пятый.Фазовые равновесия
Г л а в а 13. Термодинамическая теория фазовых равновесий 415
§ 13.1. Основные понятия 415
§ 13.2. Условия фазового равновесия 418
§ 13.3. Правило фаз Гиббса 419
Глава 14. Однокомпонентные системы 421
§ 14.1. Применение правила фаз Гиббса к однокомпонентным системам 421
§ 14.2. Фазовые переходы первого и второго рода 422
§ 14.3. Уравнение Клапейрона - Клаузиуса 425
§ 14.4. Давление насыщенного пара 423
§ 14.5. Диаграммы состояния однокомпонентных систем 429
§ 14.6. Диаграмма состояния диоксида углерода 431
§ 14.7. Диаграмма состояния воды 432
§ 14.8. Диаграмма состояния серы 433
§ 14.9. Энантиотропные и монотропные фазовые переходы 435
Г л а в а 15. Двухкомпонентные системы 436
§ 15.1. Метод физико-химического анализа 436
§ 15.2. Применение правила фаз Гиббса к двухкомпонентным системам 437
§ 15.3. Равновесие газ - жидкий раствор в двухкомпонентных системах 438
§ 15.4. Равновесие жидкость - жидкость в двухкомпонентных системах 442
§ 15.5. Равновесие пар - жидкий раствор в двухкомпокентьых системах 444
§ 15.6. Физико-химические основы перегонки растворов 453
§ 15.7. Равновесие кристаллы - жидкий раствор в двухкомпонентных системах 457
§ 15.8. Равновесие жидкость - газ и кристаллы - газ (пар) в двухкомпонентных
системах 476
§ 15-9. Расчеты по диаграммам состояния 476
Г л а в а 16. Трехкомпонентные системы 482
§ 16.1. Применение правила фаз Гиббса к трехкомпонентным системам 482
§ 16.2. Графическое изображение состава трехкомпонентной системы 482
§ 16.3. Равновесие кристаллы - жидкий раствор в трехкомпонентных системах 484
§ 16.4. Равновесие жидкость - жидкость в трехкомпонентных системах 489
§ 16.5. Распределение растворяемого вещества между двумя жидкими фазами.
Экстракция 491
Приложение 495
Предметный указатель 497
ОГЛАВЛЕНИЕ Книга 2.
Предисловие 3
Раздел шестой. Электрохимия
Г л а в а 17. Растворы, электролитов 4
§ 17.1. Предмет электрохимии 4
§ 17.2. Специфика растворов электролитов 5
§ 17.3. Электролитическая диссоциация в растворе 6
§ 17.4. Средняя ионная активность и коэффициент активности 10
§ 17.5. Основные понятия электростатической теории сильных электролитов Дебая и
Хюккеля 13
§ 17.6. Основные понятия теории ассоциации ионов 22
§ 17.7. Термодинамические свойства ионов 24
§ 17.8. Термодинамика ионной сольватации 28
Г л а в а 18. Неравновесные явления в электролитах. Электрическая
проводимость электролитов 30
§ 18.1. Основные понятия. Законы Фарадея 30
§ 18.2. Движение ионов в электрическом поле. Числа переноса ионов. 32
§ 18.3. Электрическая проводимость электролитов. Удельная электрическая
проводимость 37
§ 18.4. Электрическая проводимость электролитов. Молярная электрическая
проводимость 39
§ 18.5. Молярная электрическая проводимость ионов гидроксония и гидроксида 43
§ 18.6. Электрическая проводимость неводных растворов 44
§ 18.7. Электрическая проводимость твердых и расплавленных электролитов 46
§ 18.8. Кондуктометрия 47
Г л а в а 19. Равновесные электродные процессы 49
§ 19.1. Основные понятия 49
§ 19.2. ЭДС электрохимической системы. Электродный потенциал 51
§ 19.3. Возникновение скачка потенциала на границе раствор-металл 53
§ 19.4. Диффузионный потенциал 55
§ 19.5. Строение двойного электрического слоя на границе раствор-металл 56
§ 19.6. Термодинамика обратимых электрохимических систем 60
§ 19.7. Классификация обратимых электродов 64
§ 19.8. Электродные потенциалы в неводных растворах 74
§ 19.9. Электрохимические цепи 75
§ 19.10. Применение теории электрохимических систем к изучению равновесия в
растворах 82
§ 19.11. Потенциометрия 85
Раздел седьмой. Кинетика химических реакций
Г л а в а 20. Законы химической кинетики 93
§ 20.1. Общие понятия и определения 93
§ 20.2. Скорость химической реакции 95
§ 20.3. Закон действующих масс и принцип независимости протекания реакций 101
Г л а в а 21. Кинетика химических реакций в закрытых системах. 105
§ 21.1. Односторонние реакции первого порядка 105
§ 21.2. Односторонние реакции второго порядка 109
§ 21.3. Односторонние реакции n-го порядка 111
§ 21.4. Методы определения порядка реакции 112
§ 21.5. Двусторонние реакции первого порядка 113
§ 21.6. Двусторонние реакции второго порядка 116
§ 21.Т. Параллельные односторонние реакции 117
§ 21.8. Односторонние последовательные реакции 119
§ 21.9. Метод квазистационарных концентраций 125
Г л а в а 22. Кинетика реакций в открытых системах 127
§ 22.1. Кинетика реакций в реакторе идеального смешения 127
§ 22.2. Кинетика реакций в реакторе идеального вытеснения 129
Г л а в а 23. Теория элементарного акта химического взаимодействия 133
§ 23.1. Элементарный химический акт 133
§ 23.2. Теория активных соударений 137
§ 23.3. Теория активированного комплекса 141
§ 23.4. Предэкспоненциальный множитель в уравнении Аррениуса по теории
переходного состояния 154
§ 23.5. Симметрия МО и энергия активации химических реакций 159
Г л а в а 24. Кинетика реакций в растворах, цепные и фотохимические реакции
166
§ 24.1. Особенности кинетики реакций в растворах 166
§ 24.2. Влияние среды на константу скорости реакции 170
§ 24.3. Кинетика ионных реакций в растворах 178
§ 24.4. Цепные реакции 181
§ 24.5. Фотохимические реакции 189
Г л а в а 25. Кинетика электродных процессов 196
§ 25.1. Скорость электрохимической реакции. Ток обмена 196
§ 25.2. Электродная поляризация 197
§ 25.3. Диффузионное перенапряжение 199
§ 25.4. Электрохимическое перенапряжение 205
§ 25.5. Другие виды перенапряжения 210
5 25.6. Температурно-кинетический метод определения природы поляризации при
электрохимических процессах 211
§ 25.7. Перенапряжение при электролитическом выделении водорода 213
§ 25.8. Электролиз. Напряжение разложения 217
§ 25.9. Поляризационные явления в химических источниках электрического тока 220
§ 25.10. Электрохимическая коррозия металлов. Пассивность металлов. Методы
защиты от коррозии 222
Раздел восьмой. Катализ
Г л а в а 26. Принципы каталитическою действия 228
§ 26.1. Основные понятия и определения 228
§ 26.2. Особенности кинетики каталитических реакций 232
§ 26.3. Энергия активации каталитических реакций 237
§ 26.4. Взаимодействие реагентов с катализатором и принципы каталитического
действия 241
Г л а в а 27. Гомогенный катализ 245
§ 27.1. Кислотно-основный катализ 246
§ 27.2. Окислительно-восстановительный катализ 255
§ 27.3. Ферментативный катализ 260
§ 27.4. Автокатализ, ингибирование и периодические каталитические реакции 266
§ 27.5. Применение в промышленности и перспективы развития гомогенного катализа
271
Г л а в а 28. Гетерогенный катализ. 273
§ 28.1. Структура поверхности гетерогенных катализаторов 273
§ 28.2. Адсорбция как стадия гетерогенно-каталитических реакций 277
§ 28.3. Механизм гетерогенно-каталитических реакций 282
§ 28.4. Кинетика гетерогенно-каталитических реакций на равнодоступной
поверхности 285
§ 28.5. Макрокинетика гетерогенно-каталитических процессов 292
§ 28.6. Применение гетерогенного катализа в промышленности 300
Литература 303
Приложение 305
Предметный указатель 312
Оглавление 316
ФИЗИЧЕСКАЯ ХИМИЯ
Предмет физической химии. Её значение
Взаимосвязь химических и физических явлений изучает физическая химия. Эта отрасль химии является пограничной между химией и физикой. Пользуясь теоретическими и экспериментальными методами обеих наук, а также своими собственными методами, физическая химия занимается многосторонним исследованием химических реакций и сопутствующих им физических процессов. Поскольку, однако, даже многостороннее исследование никогда не является полным и не охватывает явление исчерпывающим образом, постольку законы и закономерности физической химии, как и других естественных наук, всегда упрощают явление и не отражают его полностью.
Быстрое развитие и растущее значение физической химии связаны с её пограничным положением между физикой и химией. Основная общая задача физической химии – предсказание временнóго хода процесса и конечного результата (состояния равновесия) в различных условиях на основании данных о строении и свойствах веществ, составляющих изучаемую систему.
Краткий очерк истории развития физической химии
Термин «физическая химия» и определение этой науки впервые были даны М.В.Ломоносовым, который в 1752-1754 гг. читал студентам Академии наук курс физической химии и оставил рукопись этого курса «Введение в истинную физическую химию» (1752). Ломоносов выполнил многие исследования, темы которых соответствуют составленному им «Плану к курсу физической химии» (1752) и программе экспериментальных работ «Опыт физической химии» (1754). Под его руководством проводился также студенческий практикум по физической химии.
Ломоносов дал следующее определение физической химии: «Физическая химия есть наука, объясняющая на основании положений и опытов физики то, что происходит в смешанных телах при химических операциях». Это определение близко к современному.
Для развития физической химии огромное значение имело открытие двух законов термодинамики в середине XIX века (С.Карно, Ю.Р.Майер, Г.Гельмгольц, Д.П.Джоуль, Р.Клаузиус, В. Томсон).
Количество и разнообразие исследований, лежащих в области, пограничной между физикой и химией, постоянно возрастало в XIX веке. Было развито термодинамическое учение о химическом равновесии (К.М.Гульдберг, П.Вааге, Д.У.Гиббс). Исследования Л.Ф.Вильгельми положили начало изучению скоростей химических реакций (химическая кинетика). Исследовался перенос электричества в растворах (И.В.Гитторф, Ф.В.Г.Кольрауш), изучались законы равновесия растворов с паром (Д.П.Коновалов) и развивалась теория растворов (Д. И. Менделеев).
Признание физической химии как самостоятельной науки и учебной дисциплины выразилось в учреждении в Лейпцигском университете (Германия) в 1887 году первой кафедры физической химии во главе с В.Оствальдом и в основании там же первого научного журнала по физической химии. В конце XIX века Лейпцигский университет был центром развития физической химии, а ведущими физико-химиками являлись В.Оствальд, Я.Х.Вант-Гофф, С.Аррениус и В.Нернст. К этому времени определились три основных раздела физической химии – химическая термодинамика, химическая кинетика и электрохимия.
К важнейшим направлениям науки, развитие которых является необходимым условием технического прогресса, относится исследование химических процессов; физической химии принадлежит ведущая роль в развитии этой проблемы.
Разделы физической химии. Методы исследования
Химическая термодинамика. В этом разделе на основе законов общей термодинамики излагаются законы химического равновесия и учение о фазовых равновесиях.
Учение о растворах ставит своей целью объяснение и предсказание свойств растворов (гомогенных смесей нескольких веществ) на основании свойств веществ, составляющих раствор.
Учение о поверхностных явлениях. Изучаются разнообразные свойства поверхностных слоёв твёрдых тел и жидкостей (границы раздела между фазами); одно из основных изучаемых явлений в поверхностных слоях – это адсорбция (накопление вещества в поверхностном слое).
В системах, где поверхности раздела между жидкими, твёрдыми и газообразными фазами сильно развиты (эмульсии, туманы, дымы и т. д.), свойства поверхностных слоёв приобретают основное значение и определяют многие своеобразные свойства всей системы в целом. Такие дисперсные (микрогетерогенные) системы изучаются коллоидной химией, которая является крупным самостоятельным разделом физической химии.
Приведенный перечень основных разделов физической химии не охватывает некоторых областей и более мелких разделов этой науки, которые можно рассматривать как части более крупных разделов или как самостоятельные разделы физической химии. Следует ещё раз подчеркнуть тесную взаимосвязь различных разделов физической химии. При исследовании любого явления приходится использовать арсенал представлений, теорий и методов исследования многих разделов химии (а нередко и других наук). Лишь при начальном знакомстве с физической химией можно в учебных целях распределить материал по указанным разделам.
Методы физико-химического исследования . Основные методы физической химии, естественно, являются методами физики и химии. Это – прежде всего экспериментальный метод – исследование зависимости свойств веществ от внешних условий, экспериментальное изучение законов протекания различных процессов и законов химического равновесия.
Теоретическое осмысление экспериментальных данных и создание стройной системы знаний основано на методах теоретической физики.
Термодинамический метод, являющийся одним из них, позволяет количественно связывать различные свойства вещества («макроскопические» свойства) и рассчитывать одни из этих свойств на основании опытных величин других свойств.
ГЛАВА I.
ПЕРВЫЙ ЗАКОН ТЕРМОДИНАМИКИ
Теплота и работа
Изменения форм движения при переходе его от одного тела к другому и соответствующие превращения энергии весьма разнообразны. Формы же самого перехода движения и связанных с ним переходов энергии могут быть разбиты на две группы.
В первую группу входит только одна форма перехода движения путём хаотических столкновений молекул двух соприкасающихся тел, т.е. путём теплопроводности (и одновременно путём излучения). Мерой передаваемого таким способом движения является теплота .
Во вторую группу включаются различные формы перехода движения, общей чертой которых является перемещение макроскопических масс под действием каких-либо внешних сил, имеющих направленный характер. Таковы поднятие тел в поле тяготения, переход некоторого количества электричества от большего электростатического потенциала к меньшему, расширение газа, находящегося под давлением и т.д. Общей мерой передаваемого такими способами движения является работа .
Теплота и работа характеризуют качественно и количественно две различные формы передачи движения от одной части материального мира к другой.
Передача движения есть своеобразное сложное движение материи, две основные формы которого мы различаем. Теплота и работа являются мерами этих двух сложных форм движения материи, и их следует рассматривать как виды энергии.
Общим свойством теплоты и работы является то, что они имеют значение только в течение отрезков времени, в которые протекают эти процессы. В ходе таких процессов в одних телах уменьшается движение в тех или иных формах и убывает соответствующая энергия, одновременно в других телах увеличивается движение в тех же или других формах и возрастают соответствующие виды энергии.
Мы не говорим о запасе теплоты или работы в каком-либо теле, а только о теплоте и работе известного процесса. После его окончания о наличии в телах теплоты или работы говорить не приходится.
Внутренняя энергия
Для некругового процесса равенство (I, 1) не соблюдается, так как система не возвращается в исходное состояние. Вместо этого равенства для некругового процесса можно записать (опуская коэффициент k ):
Так как пределы интегрирования в общем случае произвольны, то и для элементарных величин dW и dQ :
dQ ¹ dW ,
следовательно:
dQ – dW ¹ 0
Обозначим разность dQ – dW для любого элементарного термодинамического процесса через dU:
dU º dQ – dW (I, 2)
или для конечного процесса:
– (I, 2а)
Возвращаясь к круговому процессу, получаем (из уравнения I, 1):
= – = 0 (I, 3)
Таким образом, величина dU является полным дифференциалом некоторой функции состояния системы. При возвращении системы к исходному состоянию (после циклического изменения) величина этой функции приобретает первоначальное значение.
Функция состояния системы U, определяемая равенствами (I, 2) или (I, 2а), называется внутренней энергией системы .
Очевидно, выражение (I, 2а) может быть записано следующим образом:
= U 2 – U 1 = ∆U = – (I, 2б)
U 2 – U 1 = ∆U = Q – W
Данное рассуждение обосновывает опытным путем наличие определенной функции состояния системы, имеющей смысл суммарной меры всех движений, которыми система обладает.
Иначе говоря, внутренняя энергия включает поступательную и вращательную энергию молекул, колебательную энергию атомов и групп атомов в молекуле, энергию движения электронов, внутриядерную и другие виды энергии, т. е. совокупность всех видов энергии частиц в системе за исключением потенциальной и кинетической энергии самой системы.
Предположим, что циклический процесс удалось провести так, что после того, как система вернулась к исходному состоянию, внутренняя энергия системы не приняла начального значения, а увеличилась. В этом случае повторение круговых процессов вызвало бы накопление энергии в системе. Создалась бы возможность превращения этой энергии в работу и получения таким путем работы не за счёт теплоты, а «из ничего», так как в круговом процессе работа и теплота эквивалентны друг другу, что показано прямыми опытами.
Невозможность осуществления указанного цикла построения вечного двигателя (перпетуум мобиле) первого рода, дающего работу без затраты эквивалентного количества другого вида энергии, доказана отрицательным результатом тысячелетнего опыта человечества. Этот результат приводит к тому же выводу, который в частной, но более строгой форме мы получили, анализируя опыты Джоуля.
Сформулируем ещё раз полученный результат. Полный запас энергии системы (её внутренняя энергия) в результате циклического процесса возвращается к исходному значению, т. е. внутренняя энергия системы, находящейся в данном состоянии, имеет одно определенное значение и не зависит от того, каким изменениям система подвергалась перед тем, как прийти к данному состоянию.
Иными словами, внутренняя энергия системы есть однозначная, непрерывная и конечная функция состояния системы.
Изменение внутренней энергии системы определяется выражением (I, 2б); для кругового процесса справедливо выражение (I, 3). При бесконечно малом изменении некоторых свойств (параметров) системы внутренняя энергия системы изменяется также бесконечно мало. Это – свойство непрерывной функции.
В пределах термодинамики нет необходимости использовать общее определение понятия внутренней энергии. Формальное количественное определение через выражения (I, 2) или (I, 2а) достаточно для всех дальнейших термодинамических рассуждений и выводов.
Так как внутренняя энергия системы есть функция её состояния, то, как уже было сказано, прирост внутренней энергии при бесконечно малых изменениях параметров состояний системы есть полный дифференциал функции состояния. Разбивая интеграл в уравнении (I, 3) на два интеграла по участкам пути от состояния 1 до состояния 2 (путь «а») (см. рис. I) и обратно – от состояния 2 до состояния 1 (иной путь «b»), – получаем:
 (I, 4)
(I, 4)
 (I, 5)
(I, 5)
К тому же результату мы придем, сравнивая пути «а»и«с»или «b»и «с» и т. д.

Рис. I. Схема кругового (циклического) процесса.
Выражение (I, 5) показывает, что приращение внутренней энергии системы при переходе её из одного состояния в другое не зависит от пути процесса, а зависит только от начального и конечного состояния системы.
Первое начало термодинамики
Первое начало термодинамики непосредственно связано с законом сохранения энергии. Оно позволяет рассчитывать баланс энергии при протекании различных процессов, в том числе и химических реакций.
Из закона сохранения энергии следует:
Q = ∆U + W
Полученное выражение для закрытой системы может быть прочитано следующим образом: теплота, подведенная к системе, расходуется только на изменение её внутренней энергии и совершение работы.
Приведенное выше утверждение, связанное с уравнениями (I, 3) и (I, 5), служит формулировкой первого начала термодинамики (в сочетании с уравнением (I, 2), дающим количественное определение внутренней энергии).
Первое начало термодинамики является количественной формулировкой закона сохранения энергии в применении к процессам, связанным с превращениями теплоты и работы.
Еще одна формулировка первого начала термодинамики может быть получена из выражения (I, 2а). В изолированной системе dQ = 0 и dW = 0 , тогда и dU = 0 ; следовательно, при любых процессах, протекающих в изолированной системе:
![]() (I,6)
(I,6)
т. е. внутренняя энергия изолированной системы постоянна . Эта формулировка первого закона термодинамики есть примененное к конкретным условиям и конечным системам количественное выражение общего закона сохранения энергии, в соответствии с которым энергия не создается и не исчезает.
Следует отметить, что первый закон термодинамики не дает возможности найти полное значение внутренней энергии системы в каком-либо состоянии, так как уравнения, выражающие первый закон, приводят к вычислению только изменения энергии системы в различных процессах. Точно так же нельзя непосредственно измерить изменение внутренней энергии в макроскопических процессах; можно лишь вычислить это изменение с помощью уравнения (I, 2б), учитывая измеримые величины – теплоту и работу данного процесса.
Отметим, что теплота и работа (каждая в отдельности) не обладают свойством функции состояния, выражаемым уравнением (I, 3) или (I, 5) и присущим внутренней энергии. Теплота и работа процесса, переводящего систему из состояния 1 в состояние 2, зависят в общем случае от пути процесса и величины δQ и δW не являются дифференциалами функции состояния, а суть просто бесконечно малые величины, которые мы будем называть элементарной теплотой и элементарной работой.
Таким образом, дифференциал внутренней энергии dU имеет иные математические свойства, чем элементарные теплота dQ и работа dW . Это имеет существенное значение при построении системы термодинамики.
Уравнения состояния
Многие свойства системы, находящейся в равновесии, и составляющих её фаз являются взаимозависимыми. Изменение одного из них вызывает изменение других. Количественные функциональные зависимости между свойствами системы (фазы) могут быть отражены уравнениями различного вида.
Из таких уравнений наибольшее значение имеет уравнение состояния фазы, связывающее в интегральной форме давление, температуру, плотность (или объём), состав и другие свойства каждой фазы системы, находящейся в равновесии.
Уравнение состояния тесно связано с термодинамическими уравнениями системы и ее однородных частей (фаз), но не может быть в конкретной форме выведено из основных уравнений термодинамики и должно быть найдено опытным путем или получено методами статистической физики, исходя из молекулярных параметров (т. е. величин, характеризующих строение и свойства отдельных молекул). Простейшими уравнениями состояния являются уравнения для газов при малых давлениях: уравнение Клапейрона – Менделеева, уравнение Ван-дер-Ваальса и др.
Наличие уравнений состояния и других уравнений, связывающих различные свойства фазы, приводит к тому, что для однозначной характеристики состояния системы оказывается достаточным знание только нескольких, немногих независимых свойств. Эти свойства называются независимыми переменными или параметрами состояния системы. Остальные свойства являются функциями параметров состояния и определяются однозначно, если заданы значения последних. При этом для многих задач не имеет значения, известны ли нам конкретные уравнения состояния исследуемых фаз; важно только, что соответствующие зависимости всегда реально существуют.
Таким образом, состояние системы определяется независимыми переменными (параметрами состояния), число которых зависит от характера конкретной системы, а выбор их в принципе произволен и связан с соображениями целесообразности. Для определения состояния простейших систем – однородных и постоянных во времени по массе и составу (состоящих из одной фазы и не изменяющихся химически) – достаточно знать две независимые переменные из числа трех (объём V, давление P и температура T). В более сложных системах в число независимых переменных могут входить концентрации, электрический заряд, электростатический потенциал, напряженность магнитного поля и другие.
Калорические коэффициенты
Внутренняя энергия системы, будучи функцией состояния, является функцией независимых переменных (параметров состояния) системы.
В простейших системах
U = f (V, T ) (I, 7)
откуда полный дифференциал U:
dU = dV + dT (1,8)
Подставив значение dU из уравнения (I, 8) в уравнение (I, 2), находим:
δQ = dV + dT + δW (I, 9)
Если в изучаемой системе имеет место только работа расширения и отсутствуют работы электрическая, силы тяготения, поверхностных сил и т. д., то dW = PdV. Тогда
δQ = + P dV + dT (I, 9а)
Обозначив коэффициенты при дифференциалах независимых переменных в уравнении (I, 9а) символами l и C V , получим:
δQ = ldV + C V dT (1,10)
Из уравнений (I, 9а) и (I, 10) следует:
= l = + P (I,11)
= C V =
Величины и не представляют собой производных какой-либо функции. Первая из них является теплотой изотермического расширения тела. Эта величина, размерность которой совпадает с размерностью давления, складывается из внешнего давления и члена ; который отражает взаимное притяжение молекул. Этот член мал для реальных газов и очень велик (по сравнению с обычными значениями внешнего давления) для жидкостей и твердых тел.
Величина C V , в соответствии с уравнением (I, 11), есть теплоемкость при постоянном объёме . Теплота, полглощаемая системой при постоянном объёме, затрачивается полностью на увеличение внутренней энергии (при условии отсутствия всех видов работы, в том числе работы расширения).
Коэффициенты полного дифференциала внутренней энергии при переменных V и Т имеют простой физический смысл, как показано выше.
Выбрав в качестве независимых переменных P и Т или V и P и считая внутреннюю энергию функцией этих пар переменных, можно аналогично изложенному получить:
dQ = hdP + C P dT (I, 10а)
dQ = cdV + ldp (I, 10б)
где величины h, C P , c и l связаны с производными внутренней энергии более сложными соотношениями, чем представленные в уравнении (I, 11). Отметим, что C p = есть теплоемкость при постоянном давлении, а h = – теплота изотермического возрастания давления. Последняя величина существенно отрицательна.
Коэффициенты l , h, C V , C P , cи λ называются калорическими коэффициентами. Имея самостоятельный физический смысл (особенно C P , C V и l ), они являются также полезными вспомогательными величинами при термодинамических выводах и расчетах.
Работа различных процессов
Под названием работы объединяются многие энергетические процессы; общим свойством этих процессов является затрата энергии системы на преодоление силы, действующей извне. К таким процессам относится, например, перемещение масс в потенциальном поле. Если движение происходит против градиента силы, то система затрачивает энергию в форме работы; величина работы положительна. При движении по градиенту силы система получает энергию в форме работы извне; величина работы отрицательна. Такова работа поднятия известной массы в поле тяготения. Элементарная работа в этом случае:
dW = – mgdH
где m – масса тела; H – высота над начальным нулевым уровнем. При расширении системы, на которую действует внешнее давление P, система совершает работу , элементарная работа равна в этом случае PdV (V 1 и V 2 – начальный и конечный объёмы системы соответственно).
При движении электрического заряда q в электрическом поле против направления падения потенциала j и на участке, где изменение потенциала равно dj, а также при увеличении заряда тела, имеющего потенциал j , на величину dq работа совершается над системой, величина ее равна в первом случае – qdj , а во втором случае – jdq .
Аналогичным образом можно выразить работу увеличения поверхности раздела S
между однородными частями системы (фазами): dW
= -sdS
,
где s – поверхностное натяжение.
В общем случае элементарная работа dW является суммой нескольких качественно различных элементарных работ:
dW = Pd V – mgdH – sdS – jd q + … (1,12)
Здесь P, -mg, - σ, -j – силы в обобщенном смысле (обобщенные силы) или факторы интенсивности; V, H, S , q – обобщенные координаты или факторы емкости.
В каждом конкретном случае следует определить, какие виды работы возможны в исследуемой системе, и, составив соответствующие выражения для dW , использовать их в уравнении (I, 2а). Интегрирование уравнения (I, 12) и подсчет работы для конкретного процесса возможны только в тех случаях, когда процесс равновесен и известно уравнение состояния.
Для очень многих систем можно ограничить ряд уравнения (I, 12) одним членом – работой расширения.
Работа расширения при равновесных процессах выражается различными уравнениями, вытекающими из уравнения состояния. Приведем некоторые из них:
1) Процесс, протекающий при постоянном объёме (изохорный процесс; V = const ):
W = ∫δW = ∫PdV = 0 (I, 13)
2) Процесс, протекающий при постоянном давлении (изобарный процесс; P = const ):
W = = P(V 2 – V 1) = PDV (I, 14)
3) Процесс, протекающий при постоянной температуре (изотермический процесс, T = const ). Работа расширения идеального газа, для которого PV = nRT:
W = dV = nRT ln (I, 15)
Энтальпия
Уравнение первого закона термодинамики для процессов, где совершается только работа расширения, приобретает вид:
δQ = dU + PdV (I, 19)
Если процесс идет при постоянном давлении, то, интегрируя, получаем:
Q P = U 2 – U 1 + P(V 2 – V 1) (I, 20)
Q P = (U 2 + PV 2) – (U 1 + PV 1) (I, 21)
Так как P и V – параметры состояния, a U – функция состояния, то сумма U + PV такжеявляется функцией состояния и ее изменение в процессе не зависит от пути процесса, а лишь от начального и конечного состояний. Эта функция называется энтальпией и обозначается символом H . Определением величины H служит тождество:
H U + PV (I, 22)
Из уравнения (I, 21) следует, что теплота, поглощаемая при постоянном давлении, равна приросту энтальпии DH и не зависит от пути процесса:
![]() (I,21а)
(I,21а)
Второй закон термодинамики
Наиболее часто встречающимися и безусловно самопроизвольными являются процессы передачи теплоты от горячего тела к холодному (теплопроводность) и перехода работы в теплоту (трение). Многовековая житейская, техническая и научная практика человечества показали повседневную реальность этих процессов, а также невозможность самопроизвольного протекания обратных процессов, очень заманчивых с практической точки зрения (получение работы за счет отнятия теплоты у тел, окружающих рабочее тело). Это дает основание утверждать, что единственным результатом любой совокупности процессов не может быть переход теплоты от менее нагретого тела к более нагретому (постулат Клаузиуса).
Обратный указанному переход теплоты от более нагретого тела к менее нагретому – это обычный неравновесный процесс передачи теплоты путем теплопроводности. Он не может быть обращен, т. е. проведен в обратном направлении через ту же последовательность состояний. Но этого мало: если в системе прошел процесс прямой передачи теплоты, то никаким образом нельзя осуществить такую последовательность любых процессов, в результате которой все тела, участвовавшие в передаче теплоты, пришли бы в исходное состояние и не произошло бы никаких изменений в других телах. Процесс теплопроводности необратим.
Другое общее положение, имеющее ту же опытную основу, утверждает следующее: единственным результатом любой совокупности процессов не может быть превращение теплоты в работу (т. е. поглощение системой теплоты из окружающей среды и отдача эквивалентной этой теплоте работы). Таким образом, самопроизвольный процесс превращения работы в теплоту (путем трения) необратим (так же, как и теплопроводность).
Последнее утверждение может быть изложено иначе: теплота наиболее холодного из участвующих в процессе тел не может служить источником работы (постулат Томсона).
Оба положения (постулаты Клаузиуса и Томсона) являются формулировками второго закона термодинамики и эквивалентны друг другу, т. е. каждое из них может быть доказано на основании другого.
Так как переход теплоты или её превращение в работу рассматривается как единственный результат процесса, то очевидно необходимо, чтобы система, участвующая в теплообмене, возвращалась в результате процесса или совокупности процессов в первоначальное состояние. При таком циклическом процессе внутренняя энергия системы не изменится.
Предположим, что вторая из приведенных выше формулировок (особенно в последней ее форме) неправильна. Тогда можно было бы построить машину, работающую циклами, «рабочее тело» которой периодически возвращалось бы в исходное состояние, причем эта машина давала бы работу за счёт теплоты, поглощаемой извне от тела, не более нагретого, чем сама система и все другие окружающие систему тела. Такой процесс протекал бы без нарушения первого закона термодинамики (работа за счет теплоты), но для практики он равноценен получению работы из ничего, так как всякая машина имела бы практически неисчерпаемый источник теплоты в окружающей среде. Так пароход мог бы двигаться, отнимая теплоту океанской воды и не нуждаясь в топливе. Такая машина называется перпетуум мобиле (вечный двигатель) второго рода. Исходя из этого определения, можно сформулировать второй закон термодинамики, придав постулату Томсона иную форму: перпетуум мобиле второго рода невозможен.
Следует подчеркнуть, что как положения Клаузиуса и Томсона, так и утверждение о невозможности перпетуум мобиле второго рода не доказываются на основании других законов или положений. Они являются предположениями, которые оправдываются всеми следствиями, из них вытекающими, но не могут быть доказаны для всех возможных случаев.
Приведем еще одну формулировку второго закона термодинамики, являющуюся, безусловно, достаточно точной и краткой. В этой формулировке содержится постулат о существовании новой функции состояния, через которую выражается различие между обратимыми и необратимыми процессами:
Методы расчета энтропии
Уравнения (II, 1) и (II, 1а), определяющие энтропию, являются единственными исходными уравнениями для термодинамического расчета изменения энтропии системы. Заменяя элементарную теплоту в уравнении (II, 1а) ее выражениями через калорические коэффициенты (см. уравнения (I, 10) и (I, 10а)), получаем для равновесных процессов:
КДж/моль; температура плавления t пл. = 5,5°С (Т = 278,5 К ). Следовательно, изменение энтропии 1 моль бензола при плавлении (энтропия плавления) равно:
DS пл. = 35,06 Дж/моль
2. Нагревание при постоянном давлении (изобарный процесс; P = const ). Из уравнений (I, 18а) и (II, 1а)получаем:
DS = (II, 6)
Найдем изменение энтропии одного моля алюминия при нагревании от 25 до 600°С. Истинная мольная теплоемкость алюминия может быть выражена уравнением:
С р = 565,5 + 0,290 Т. По уравнению (II, 6) изменение энтропии будет равно:
DS = = 565,5 + 0,290(873 – 298) = 607,8 + 166,8 = 774,6 Дж/мольK
Постулат Планка. Абсолютные значения энтропии
По уравнению (II, 3) невозможно вычислить абсолютное значение энтропии системы. Такую возможность дает новое, недоказуемое положение, не вытекающее из двух законов термодинамики, которое было сформулировано М.Планком (1912). Согласно этому положению, называемому постулатом Планка , энтропия индивидуального кристаллического вещества при абсолютном нуле равна нулю :
Строго говоря, постулат Планка справедлив только для индивидуальных веществ, кристаллы которых идеально построены (в кристаллической решетке все узлы заняты молекулами или атомами, правильно чередующимися и закономерно ориентированными). Такие кристаллы называются идеальными твердыми телами. Реальные кристаллы не являются таковыми, так как их кристаллическая решетка построена не идеально.
Энтропия кристаллической решетки, построенной в некоторой степени беспорядочно, больше энтропии идеально построенной кристаллической решётки. Поэтому реальные кристаллы и при 0 К обладают энтропией, большей нуля. Однако энтропии реальных хорошо образованных кристаллов индивидуальных веществ при абсолютном нуле невелики.
В соответствии с постулатом Планка уравнение (II, 6) для идеального твёрдого тела примет вид:
Постулат Планка используется при термодинамическом исследовании химических процессов для вычисления абсолютных значений энтропии химических соединений – величин, которые имеют большое значение при расчете химических равновесий.
Энтропия широко используется в технической термодинамике (теплотехнике), как один из важных параметров рабочего тела в тепловой машине, например, водяного пара. Величины энтропии водяного пара в данном состоянии вычисляются по сравнению с некоторым стандартным состоянием – обычно 0°С и 1 amм. Эти значения энтропии используются для построения так называемых энтропийных диаграмм состояния водяного пара в координатах S-Т или S-H (диаграмма Молье). В таких диаграммах подобно диаграммам V-P можно изображать различные процессы, протекающие в рабочем теле тепловой машины и составляющие рабочие циклы машины.
В заключение следует отметить, что нам не придется углубляться в область термодинамики. Наша цель лишь проиллюстрировать основные идеи этой науки и объяснить причины, по которым возможно основываться на её аргументах.
Наконец, два закона термодинамики часто формулируют так:
Первый закон: Энергия Вселенной всегда постоянна.
Второй закон: Энтропия Вселенной всегда возрастает.
Физ. химия - наука о закономерностях хим.процессов и хим. явлений.
Предмет физ.химии объяснение хим. явлений на основе более общих законов физики. Физ.химия рассматривает две основные группы вопросов:
1. Изучение строения и свойств вещества и составляющих его частиц;
2. Изучение процессов взаимодействия веществ.
Физ.химия ставит целью изучение связей м/у хим-ми и физ-ми явлениями. Знание таких связей необходимо для того, чтобы глубже изучить хим.реакции, протекающие в природе и используемые в технолог. процессах, управлять глубиной и направлением реакции. Основной целью дисциплины Физ.химия изучение общих связей и закономерностей хим. процессов, основанных на фундаментальных принципах физики. Физ.химия применяет физ. теории и методы к хим.явлениям.
Она объясняет ПОЧЕМУ и КАК происходят превращения веществ: хим. реакции и фазовые переходы. ПОЧЕМУ – хим.термодинамика. КАК- химическая кинетика.
Основные понятия физ.химии
Основной объект хим. термодинамики –это термодинамическая система. Термодинамич. система – любое тело или совокупность тел, способных обмениваться м/у собой и с др. телами энергией и в-вом. Системы подразделяют на открытые, закрытые и изолированные. Открыт ая - термодинамическая система обменивается с внешней средой и в-вом и энергией. Закрыт ая -система, в которой отсутствует обмен в-вом с окружающей средой, но она может обмениваться с ней энергией. Изолированн ая -система объем остается постоянным и лишена возможности обмениваться с окружающей средой и энергией и в-вом.
Система может быть гомогенной (однородной) или гетерогенной (неоднородной ). Фаза - это часть системы, которая в отсутствии внешнего поля сил обладает одинаковым составом во всех своих точках и одинаковыми термодинамич. св-вами и отделена от других частей системы поверхностью раздела. Фаза всегда однородна, т.е. гомогенна, поэтому однофазная система называется гомогенной. Система, состоящая из неск-ких фаз, называется гетерогенной.
Свойства системы подразделить на две группы: экстенсивные и интенсивные.
В термодинамике используются понятия равновесных и обратимых процессов. Равновесным –это процесс, проходящий через непрерывный ряд состояний равновесия. Обратимый термодинамический процесс – это процесс, который может быть проведен в обратном направлении без того, чтобы в системе и окружающей среде остались какие-либо изменения.
2. I-ый закон термодинамики. Внутренняя энергия, теплота, работа.
Первое начало термодинамики непосредственно связано с законом сохранения энергии. Исходя из этого закона, следует, что в любой изолированной системе запас энергии остается постоянным. Из законасохранения энергии вытекает еще одна формулировка первого начала термодинамики – невозможность создания вечного двигателя (perpetuum mobile) первого рода, который производил бы работу, не затрачивая на это энергии. Особенно важной для химической термодинамики формулировкой
первого начала является выражение его через понятие внутренней энергии: внутренняя энергия является функцией состояния, т.е. её изменение не зависит от пути процесса, а зависит только от начального и конечного состояния системы. Изменение внутренней энергии системы U может происходить за счет обмена теплотой Q и работой W с окружающей средой. Тогда из закона сохранения энергии следует, что полученная системой извне теплота Q расходуется на приращение внутренней энергии ΔU и работу W, совершенную системой, т.е. Q = ΔU +W . Данное у равнение является
математическим выражением первого начала термодинамики.
I начало термодинамики его формулировки:
в любой изолированной системе запас энергии остается постоянным;
разные формы энергии переходят друг в друга в строго эквивалентных количествах;
вечный двигатель (perpetuum mobile ) первого рода невозможен;
внутренняя энергия является функцией состояния, т.е. её изменение не зависит от пути процесса, а зависит только от начального и конечного состояния системы .
аналитическое выражение: Q = D U + W ; для бесконечно малого изменения величин d Q = dU + d W .
1-ое начало термодинамики устанавливает соотнош. м/у теплотой Q, работой А и изменением внутр. энергии системы ΔU. Изменение внутр. энергии системы равно кол-ву сообщенной системе теплоты минус кол-во работы, совершенной системой против внешних сил.
Уравнение (I.1)- математическая запись 1-го начала термодинамики, уравнение (I.2) – для бесконечно малого изменения сост. системы.
Внутр. энергия- функция сост.; это означает, что измен-е внутр. энергии ΔU не зависит от пути перехода системы из состояния 1 в состояние 2 и равно разности величин внутр. энергии U2 и U1 в этих состояниях: (I.3)
Внутр. энергия системы- это сумма потенциальной энергии взаимодейст. всех частиц тела м/у собой и кинетической энергии их движения (без учета кинетич. и потенциальн. энергий системы в целом). Внут. энергия системы зависит от природы в-ва, его массы и от параметров состоянии системы. Она возраст. с увеличением массы системы, так как является экстенсивным св-вом системы. Внутр. энергию обозначают литерой U и выражают в джоулях (Дж). В общем случае для системы с кол-вом в-ва 1 моль. Внутр. энергия, как и любое термодинамич. св-во системы, явл-ся функцией сост. Непосредственно в эксперименте проявляются только изменения внутр. энергии. Именно поэтому при расчетах всегда оперируют с её изменением U2 –U1 = U.
Все изменения внутр. энергии делятся на две группы. В 1-ую группу входит только 1-а форма перехода движения путем хаотических столкновений молекул двух соприкасающихся тел, т.е. путём теплопроводности (и одновременно путём излучения). Мерой передаваемого таким способом движения является теплота. Понятие теплоты связано с поведением огромного числа частиц – атомов, молекул, ионов. Они находятся в постоянном хаотическом (тепловом) движении. Теплота – форма передачи энергии. Второй способ обмена энергией – работа. Этот обмен энергии обусловлен действием, совершаемым системой, или действием, совершаемым над ней. Обычно работу обозначают символом W . Работа, также как и теплота, не является функцией состояния системы, поэтому величину, соответствующую бесконечно малой работе, обозначают символом частной производной - W .
Начало физической химии было положено в середине XVIII века . Термин «Физическая химия», в современном понимании методологии науки и вопросов теории познания , принадлежит М. В. Ломоносову , который в впервые читал студентам Петербургского университета «Курс истинной физической химии». В преамбуле к этим лекциям он даёт такое определение: «Физическая химия - наука, которая должна на основании положений и опытов физических объяснить причину того, что происходит через химические операции в сложных телах». Учёный в трудах своей корпускулярно-кинетической теории тепла касается вопросов, в полной мере отвечающих вышеизложенным задачам и методам. Именно такой характер носят и экспериментальные действия, служащие подтверждению отдельных гипотез и положений настоящей концепции. М. В. Ломоносов следовал таким принципам во многих направлениях своих исследований: в разработке и практической реализации основанной им же «науки о стекле», в различных опытах, посвящённых подтверждению закона сохранения вещества и силы (движения); - в работах и экспериментах, имеющих отношение к учению о растворах - им была разработана обширная программа исследований настоящего физико-химического феномена, находящаяся в процессе развития до настоящего времени.
Затем последовал более чем столетний перерыв и одним из первых в России физикохимические исследования в конце 1850-х годов начал Д. И. Менделеев .
Следующий курс физической химии читал уже Н. Н. Бекетов в Харьковском университете в 1865 году .
Первая в России кафедра физической химии была открыта в 1914 году на физико-математическом факультете Санкт−Петербургского университета, осенью приступил к чтению обязательного курса и практическим занятиям по физической химии ученик Д. П. Коновалова М. С. Вревский .
Первый научный журнал, предназначенный для публикации статей по физической химии, был основан в 1887 году В. Оствальдом и Я. Вант-Гоффом .
Предмет изучения физической химии
Физическая химия является основным теоретическим фундаментом современной химии, использующим теоретические методы таких важнейших разделов физики, как квантовая механика , статистическая физика и термодинамика , нелинейная динамика , теория поля и др. Она включает учение о строении вещества, в том числе: о строении молекул, химическую термодинамику , химическую кинетику и катализ . В качестве отдельных разделов в физической химии выделяют также электрохимию , фотохимию , физическую химию поверхностных явлений (в том числе адсорбцию), радиационную химию , учение о коррозии металлов , физико-химию высокомолекулярных соединений (см. физика полимеров) и др. Весьма близко примыкают к физической химии и подчас рассматриваются как её самостоятельные разделы коллоидная химия , физико-химический анализ и квантовая химия . Большинство разделов физической химии имеет достаточно чёткие границы по объектам и методам исследования, по методологическим особенностям и используемому аппарату.
Различие между физической химией и химической физикой
Молекулярность элементарной реакции - число частиц, которые, согласно экспериментально установленному механизму реакции, участвуют в элементарном акте химического взаимодействия. Мономолекулярные реакции - реакции, в которых происходит химическое превращение одной молекулы (изомеризация, диссоциация и т. д.): H 2 S → H 2 + S Бимолекулярные реакции - реакции, элементарный акт которых осуществляется при столкновении двух частиц (одинаковых или различных): СН 3 Вr + КОН → СН 3 ОН + КВr Тримолекулярные реакции - реакции, элементарный акт которых осуществляется при столкновении трёх частиц: О 2 + NО + NО → 2NО 2 Реакции с молекулярностью более трёх неизвестны. Для элементарных реакций, проводимых при близких концентрациях исходных веществ, величины молекулярности и порядка реакции совпадают. Чётко определённой взаимосвязи между понятиями молекулярности и порядка реакции нет, так как порядок реакции характеризует кинетическое уравнение реакции, а молекулярность - механизм реакции. Основными направлениями химической термодинамики являются:
Химическая термодинамика тесно соприкасается с такими разделами химии , как
Физико-химический анализВ основе теории физико-химического анализа лежат сформулированные Н. С. Курнаковым принципы соответствия и непрерывности. Принцип непрерывности утверждает, что если в системе не образуются новые фазы или не исчезают существующие, то при непрерывном изменении параметров системы свойства отдельных фаз и свойства системы в целом изменяются непрерывно. Принцип соответствия утверждает, что каждому комплексу фаз соответствует определённый геометрический образ на диаграмме состав-свойство . Теория реакционной способности химических соединенийХимия высоких энергий (ХВЭ) изучает химические реакции и превращения, происходящие в веществе под воздействием нетепловой энергии. Механизмы и кинетика таких реакций и превращений характеризуются существенно неравновесными концентрациями быстрых, возбуждённых или ионизированных частиц с энергией большей, чем энергия их теплового движения и в ряде случаев химической связи. Носители нетепловой энергии, воздействующей на вещество: ускоренные электроны и ионы, быстрые и медленные нейтроны, альфа- и бета-частицы, позитроны, мюоны, пионы, атомы и молекулы при сверхзвуковых скоростях, кванты электромагнитного излучения, а также импульсные электрические, магнитные и акустические поля. Процессы химии высоких энергий различают по временны́м стадиям на физическую, протекающую за время фемтосекунд и менее, в течение которого нетепловая энергия распределяется в среде неравномерно и образуется «горячее пятно», физико-химическую, в течение которой проявляется неравновесность и негомогенность в «горячем пятне» и, наконец, химическую, в которой превращения вещества подчиняются законам общей химии. В результате образуются такие ионы и возбуждённые состояния атомов и молекул при комнатных температурах, которые не могут возникнуть за счёт равновесных процессов. Внешним проявлением ХВЭ служит образование ионов и возбуждённых состояний атомов и молекул при комнатных температурах, при которых эти частицы не могут возникнуть за счёт равновесных процессов. Н. Е. Аблесимов сформулировал релаксационный принцип управления свойствами неравновесных физико-химических систем. В случае, когда времена релаксации много больше длительности физического воздействия, существует возможность управления выходом химических форм, фаз и, как следствие, свойствами веществ (материалов), используя сведения о механизмах релаксации в неравновесных конденсированных системах на физико-химической стадии релаксационных процессов (в том числе и в процессе эксплуатации). Основные разделы ХВЭ
и другие. Лазерная химияИонизирующей способностью обладают электромагнитные излучения (рентгеновское излучение , γ-излучение , синхротронное излучение) и потоки ускоренных частиц (электронов , протонов , нейтронов , гелионов , тяжёлых ионов; осколки деления тяжёлых ядер и др.), энергия которых превышает потенциал ионизации атомов или молекул (в большинстве случаев, лежащий в пределах 10-15 эВ). В рамках радиационной химии рассматриваются некоторые химические процессы, невозможные при использовании традиционных химических подходов. Ионизирующие излучения могут сильно снижать температуру протекания химических реакций без применения катализаторов и инициаторов. История радиационной химии Радиационная химия возникла после открытия x-лучей В. Рентгеном в 1895 году и радиоактивности А. Беккерелем в 1896 году, которые первыми наблюдали радиационные эффекты в фотопластинках. Первые работы по радиационной химии были выполнены в 1899-1903 годах супругами М.Кюри и П. Кюри . В последующие годы наибольшее число исследований было посвящено радиолизу воды и водных растворов . Ядерная химияОсновные направления ядерной химии:
ЭлектрохимияЭлектрохи́мия - раздел химической науки, в котором рассматриваются системы и межфазные границы при протекании через них электрического тока , исследуются процессы в проводниках , на электродах (из металлов или полупроводников , включая графит) и в ионных проводниках (электролитах). Электрохимия исследует процессы |
Классификация наук основана на классификации форм движения материи и их взаимосвязи и различии. Поэтому для того, чтобы наметить границы физической химии с рядом разделов физики и химии, следует рассмотреть связь и различие между химической и физической формами движения.
Для химической формы движения, т. е. для химического процесса, характерно изменение числа и расположения атомов в молекуле реагирующих веществ. Среди многих физических форм движения (электромагнитное поле, движение и превращения элементарных частиц, физика атомных ядер и др.) особенно тесную связь с химическими процессами имеет внутримолекулярная форма движения (колебания в молекуле ее электронное возбуждение и ионизация). Простейший химический процесс - элементарный акт термической диссоциации молекулы имеет место при нарастании интенсивности (амплитуды и энергии) колебаний в молекуле, особенно колебаний ядер вдоль валентной связи между ними. Достижение известной критической величины энергии колебаний по направлению определенной связи в молекуле приводит к разрыву этой связи и диссоциации молекулы на две части.
Более сложные реакции с участием нескольких (обычно двух) молекул можно рассматривать как соединение двух молекул при их столкновении в непрочный и короткоживущий комплекс (так называемый активный комплекс) и быстро наступающее разрушение этого комплекса на новые молекулы, так как этот комплекс при внутренних колебаниях оказывается неустойчивым по определенным связям.
Таким образом, элементарный химический акт является особой, критической точкой колебательного движения молекул. Последнее само по себе не может считаться химическим движением, однако оно является основой для первичных химических процессов.
Для химического превращения значительных масс вещества, т. е. множества молекул, являются необходимыми столкновение молекул и обмен энергиями между ними (перенос энергии движения молекул продуктов реакции к молекулам исходных веществ путем столкновений). Таким образом, реальный химический процесс тесно связан и со второй физической формой движения - хаотическим движением молекул макроскопических тел, которое часто называют тепловым движением.
Выше намечены кратко и в самых общих чертах взаимные отношения химической формы движения с двумя физическими формами движения. Очевидно, имеются такие же связи химического процесса с излучением движения электромагнитного поля, с ионизацией атомов и молекул (электрохимия) и т.д.
Строение вещества . Этот раздел включает в себя строение атомов, строение молекул и учение об агрегатных состояниях.
Учение о строении атомов имеет большее отношение к физике, чем к физической химии. Это учение является основой для изучения строения молекул.
В учении о строении молекул исследуются геометрия молекул, внутримолекулярные движения и силы, связывающие атомы в молекуле. В экспериментальных исследованиях строения молекул наибольшее применение получил метод молекулярной спектроскопии (включая радиоспектроскопию), широко используются также электрические, рентгенографические, магнитные и другие методы.
В учении об агрегатных состояниях рассматриваются взаимодействия молекул в газах, жидкостях и кристаллах, а также свойства веществ в различных агрегатных состояниях. Этот очень важный для физической химии раздел науки может считаться частью физики (молекулярная физика).
Весь раздел о строении вещества может рассматриваться также как часть физики.
Химическая термодинамика . В этом разделе на основе законов общей термодинамики излагаются законы химического равновесия и учение о фазовых равновесиях, которое обычно называют правилом фаз. Частью химической термодинамики является термохимия, в которой рассматриваются тепловые эффекты химических реакций.
Учение о растворах ставит своей целью объяснение и предсказание свойств растворов (гомогенных смесей нескольких веществ) на основании свойств веществ, составляющих раствор.
Решение этой задачи требует построения общей теории взаимодействия разнородных молекул, т. е. решения основной задачи, молекулярной физики. Для развития общей теории и частных обобщений изучаются молекулярная структура растворов и различные их свойства в зависимости от состава.
Учение о поверхностных явлениях . Изучаются разнообразные свойства поверхностных слоев твердых тел и жидкостей (границы раздела между фазами); одно из основных изучаемых явлений в поверхностных слоях - это адсорбция (накопление веществ в поверхностном слое).
В системах, где поверхности раздела между жидкими, твердыми и газообразными фазами сильно развиты (коллоидные растворы, эмульсии, туманы, дымы), свойства поверхностных слоев приобретают основное значение и определяют многие своеобразные свойства всей системы в целом. Такие микрогетерогенные системы изучаются коллоидной химией, которая является крупным самостоятельным разделом физической химии и самостоятельной учебной дисциплиной в химических высших учебных заведениях.
Электрохимия. Изучается взаимодействие электрических явлений и химических реакций (электролиз, химические источники электрического тока, теория электросинтеза). В электрохимию включают обычно учение о свойствах растворов электролитов, которое с равным правом можно отнести и к учению о растворах.
Химическая кинетика и катализ . Изучается скорость химических реакций, зависимость скорости реакции от внешних условий (давление, температура, электрический разряд и др.), связь скорости реакции со строением и энергетическими состояниями молекул, влияние на скорость реакции веществ, не участвующих в стехиометрическом уравнении реакции (катализ).
Фотохимия. Исследуется взаимодействие излучения и веществ, участвующих в химических превращениях (реакции, протекающие под влиянием излучения, например фотографические процессы и фотосинтез, люминесценция). Фотохимия тесно связана с химической кинетикой и учением о строении молекул.
Приведенный перечень основных разделов физической химии не охватывает некоторых недавно возникших областей и более мелких разделов этой науки, которые можно рассматривать как части более крупных разделов или как самостоятельные разделы физической химии. Таковы, например, радиационная химия, физико-химия высокомолекулярных веществ, магнетохимия, газовая электрохимия и другие разделы физической химии. Значение некоторых из них в настоящее время быстро растет.
Методы физико-химического исследования
Основные методы физической химии, естественно, являются методами физики и химии. Это - прежде всего экспериментальный метод - исследование зависимости свойств веществ от внешних условий и экспериментальное изучение законов протекания химических реакций во времени и законов химического равновесия.
Теоретическое осмысливание экспериментального материала и создание стройной системы знаний свойств веществ и законов химических реакций основано на следующих методах теоретической физики.
Квантово-механический метод (в частности, метод волновой механики), лежащий в основе учения о строении и свойствах отдельных атомов и молекул и взаимодействии их между собой. Факты, относящиеся к свойствам отдельных молекул, получаются, главным образом, с помощью экспериментальных оптических методов.
Метод статистической физики , дающий возможность рассчитать свойства вещества; состоящего из множества молекул («макроскопические» свойства), на основании сведений о свойствах отдельных молекул.
Термодинамический метод , позволяющий количественно связывать различные свойства вещества («макроскопические» свойства) и рассчитывать одни из этих свойств на основании опытных величин других свойств.
Современные физико-химические исследования в любой конкретной области характеризуются применением разнообразных экспериментальных и теоретических методов для изучения различных свойств веществ и выяснения их связи со строением молекул. Вся совокупность данных и указанные выше теоретические методы используются для достижения основной цели - выяснения зависимости направления, скорости и пределов протекания химических превращений от внешних условий и от строения молекул - участников химических реакций.
Вам также будет интересно:

ОБАМА - ЕВРЕЙ ИЛИ МУСУЛЬМАНИН? АНТИХРИСТ!
Обама кто ты?
Изменится не только Америка, Я...

Часть третья
Как Сталин стал главой государства
В терновом венке революций
Бытует мнение...

Анастасия Гредякина
Коррекционная предметно-развивающая среда логопедической группы и...

Образованные на основе бензола. При нормальных условиях представляют собой твердые ядовитые...
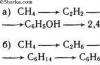
а) Из метана при нагревании можно получит ацетилен:В присутствии катализатора ацетилен...

